Congenital Anomalies
Patent Ductus Arteriosus
 MORPHOLOGY
MORPHOLOGY
Patent ductus arteriosus connects the pulmonary artery to the descending aorta in the fetal circulation, entering from the medial side, distal to the left subclavian artery. The muscle tissue of the ductus arteriosus is sensitive to prostaglandin produced by the placenta during pregnancy. After delivery, prostaglandin levels in the blood drop, resulting in muscle contraction and closure of the ductus arteriosus. In a full-term new-born babies, it closes within a few days up to 3 weeks. The wall of a persistent patent ductal arteriosus has a different histological structure - it lacks the typical contractile ability.
Shunting of the ductus arteriosus is left-to-right, that means from the aorta to the pulmonary artery, and blood flows in this direction not only in systole but also in diastole. Pulmonary flow is increased. A large left-to-right shunt increases the pressure in the pulmonary vascular bed and promotes the development of pulmonary vascular obstructive disease. Gradually, severe irreversible pulmonary hypertension develops and a shunt flow reversal to right-to-left may occur (so-called Eisenmenger syndrome). This flow is no longer typically continuous.
SYMPTOMS
A small patent ductus arteriosus can be clinically subtle until adulthood where it manifests as a murmur. In a large PDA, the left-to-right shunt flow increases with a decrease in pulmonary resistance after birth. Increased pulmonary flow causes volume overload of the left ventricle which leads to heart failure and pulmonary edema. It manifests as dyspnoea, tachypnoea and frequent respiratory infections.
With high pulmonary resistance in Eisenmenger syndrome in PDA, a cyanosis manifests only in the lower half of the body (under the entry of the ductus arteriosus into the aorta), this can even lead to right-sided cardiac decompensation.
DIAGNOSIS
On auscultation, a typical continuous (systolic-diastolic) murmur in the area under the left clavicle is present. In Eisenmenger syndrome in PDA, there is only a systolic murmur, or accentuation of the second heart sound above the pulmonary artery.
ECG is normal. With increasing hemodynamic severity of the shunt flow, signs of volume overload of the left ventricle may be observed.
On an X-ray, an enlarged heart silhouette and increased pulmonary vascular congestion can be observed.
Echocardiography is the most important modality for diagnosis. Using two-dimensional and doppler echocardiography one can determine the size, location and hemodynamic significance of the shunt.
Catheterization is performed in Eisenmenger syndrome to determine the reversibility of pulmonary hypertension. However, due to early diagnosis and surgery, we no longer see Eisenmenger's syndrome in PDA.
OPERATION
Indications for PDA closure. In premature new-borns, an attempt to pharmacologically close the shunt with indomethacin or ibuprofen (prostaglandin synthesis antagonists) is indicated. In case of failure the operation is required. In neonates and infants, the defect is operated only in case of uncontrollable heart failure (critical congenital heart defect). For asymptomatic PDA, it is advisable to carry out surgery during toddler years.
Surgery is contraindicated in Eisenmenger syndrome or in the presence of complex anomalies where blood circulation is dependent on shunt flow persistence (pulmonary atresia, Fallot tetralogy, uncorrected transposition of the great vessels, etc.). In adulthood, the operation is rare and is usually associated with a high risk as a result of increased fragility and calcification of the ductus wall, as well as the presence of pulmonary hypertension.
The first successful surgical closure of PDA in Czech Republic was done in 1947 by prof. Bedrna in Hradec Kralove.
The surgical approach is the left-sided thoracotomy. After exposure of the PDA, it is closed with 2-3 ligatures. An alternative method is resection of the duct with stitching of both stumps, which is necessary for a wide short PDA - it is rarely performed today. It is possible to close a patent ductus arteriosus by thoracoscopy (using clips) or by catheterization technique (using a spiral that fills the lumen of the entire duct).
RESULTS
The surgical risk of closure of an isolated PDA during planned operations in young children is less than 1%. It increases in premature children, in critical defects and also in rare adult operations.
The prognosis after successful PDA closure is good. If a hemodynamically insignificant PDA is not occluded, the prognosis into adulthood is good, but patients are at risk of developing endarteritis in the duct. A significant shunt is rare in adulthood. It can be solved by standard methods, but if Eisenmenger syndrome is already present, then the prognosis is poor.
Coarctation of the Aorta
 MORPHOLOGY
MORPHOLOGY
Coarctation is a narrowing of the aorta located mostly just below the left subclavian artery offset in the area of entry of the ductus arteriosus, so-called aortic isthmus. It is caused by the inner lining of the aorta bulging towards the opening of the ductus arteriosus. Narrowing of the new-born aortic lumen below 2 mm is considered tight because it constitutes a significant obstacle to blood flow in the descendent aorta, which is 5 mm wide. Below the coarctation site, the aorta is dilated.
Less frequently, the coarctation may be associated with aortic arch hypoplasia and / or a longer tubular narrowing of the isthmus. Extreme variants include interruption (termination) or atresia of the aortic arch (the entire arch is replaced by a fibrous band). Aortic coarctation is often associated with bicuspid aortic valve; other congenital heart defects such as ventricular septal defect may also be present.
SYMPTOMS
The most common manifestation of aortic coarctation is systemic hypertension in the upper body with pressure overload of the left ventricle. Through the internal thoracic arteries and intercostal arteries, a collateral circulation is created into the descending aorta, which provides adequate blood supply for the lower half of the body. If this collateral circulation is not formed (especially in new-borns), coarctation manifests as a critical defect with left heart failure and circulatory collapse. After closure of the PDA after delivery, hypoperfusion occurs in areas beyond the stenosis with renal failure, oliguria, metabolic acidosis and multiorgan failure.
Coarctation of the aorta with well-formed collateral circulation is not seen until late childhood or adulthood. Upper limb hypertension, headaches, epistaxis, weakness and tiredness of lower limbs, exertional shortness of breath and chest pain may be present. Serious complications include cerebral haemorrhage and aortic dissection or rupture.
DIAGNOSIS
Auscultation: systolic murmur above the aorta and between the shoulder blades; collaterals manifest as continuous murmur in the intercostal spaces.
Palpation: Fading or markedly weakened and delayed pulsation of the femoral arteries. Blood pressure is lower in the lower limbs compared to the upper limbs. A pressure gradient above 20 mm Hg is considered significant and indicative of coarctation.
ECG: left ventricular hypertrophy. ST-depressions and left ventricular overload signs can be suspicious of simultaneous aortic valve stenosis.
Chest X-ray: notch in the descending thoracic aorta at the site of coarctation (sign of the three - short ligament arteriosum deforms the aortic isthmus into the shape of "3), lower rib notching as a result of widened collaterals of the intercostal arteries. Cardiomegaly and pulmonary congestion may be present.
Echocardiography: suprasternal two-dimensional echocardiography evaluates the morphology of the aortic arch and the offsets of the large vessels. With Doppler, coarctation gradient can be assessed by measuring the increased systolic velocity in the descending thoracic aorta, typically there is a simultaneous diastolic forward flow at the site of coarctation.
Catheterization and aortography: determination of coarctation gradient, precise evaluation of coarctation morphology, presence of collaterals, aneurysms, abnormalities in large arteries, coronarography.
OPERATION
Indication for surgery is urgent in neonates and infants in the case of uncontrollable left heart failure. Elective surgery is best performed at the age of 3-5 years. It is performed mainly in asymptomatic children with systemic hypertension. In children without hypertension, it is indicated when a significant increase in coarctation gradient after exercise (from 10 to 60 mmHg), a significant hypertensive reaction to exercise, myocardial ischemia, or severe aortic deformity at the isthmus (in the form of the number ‘3’) are present.
Coarctation surgery is performed through left-sided thoracotomy. According to the morphological finding it is possible to choose one of many techniques to resolve the coarctation. Most preferred and probably the best technique is a complete resection of the coarctation and an end-to-end anastomosis. In the case of a longer narrowed section, a Waldhausen procedure (a subclavian flap angioplasty) is used, in which the coarctation area is longitudinally incised and widened with a flap from the ligated left subclavian artery. Another procedure, by Vosschulte, is to enlarge the coarctation site by sewing a patch in. In a case of a long pathologically altered aortic region (hypoplasia, aneurysm), it is necessary to resect the tissue and replace it with a vascular prosthesis. Bypass surgery (the aortoaortic or subclavioaortic bypass) is sometimes performed in case of recoarctation and in adults. Interventional treatment (dilation, stenting) comes also into consideration.
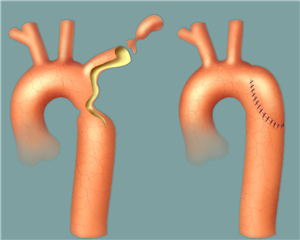
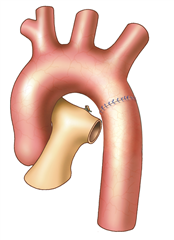
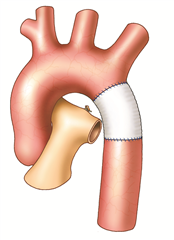

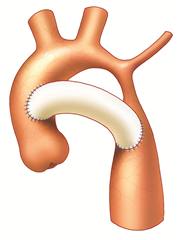

RESULTS
Surgical mortality is low. Operative and postoperative complications are rare but very serious (bleeding from the suture areas or from fragile collaterals, paradoxical postoperative hypertension with bowel paresis, spinal cord ischemia with paraplegia of the lower limbs, pseudoaneurysm around the vascular prosthesis, persistence of hypertension in patients operated late). Some patients may develop a recoarctation (up to 20% risk). In the case of hemodynamically significant recoarctation, balloon percutaneous angioplasty, possibly in combination with a metal stent implantation is currently possible. If this method fails, we proceed to re-do surgery.
Atrial Septal Defect
 MORPHOLOGY
MORPHOLOGY
Atrial septal defect (ASD) refers to a pathological communication between the right and left atria. The defect allows pathological blood shunting between the atria. The shunt direction is left-to-right due to greater compliance of the right atrium and right ventricle. There is an increased pulmonary flow, dilatation and volume overload of the right ventricle and pulmonary artery, eventually the right atrium (in presence of tricuspid valve regurgitation). The left ventricle remains normal. The ratio of increased pulmonary blood flow to systemic flow (Qp: Qs) is a quantification method. Atrial defects usually do not spontaneously close.
The defect can be located in different areas of the interatrial septum. The location of the defect is determined by its embryonic origin and determines the surgical technique of closure.
- Ostium secundum ASD (type II defect, fossa ovalis defect).
The most common type (about 70% of all atrial-level defects). The foramen ovale which is an important structure for right-to-left shunt in fetal circulation, closes in the first week after birth by sealing the two folds that form it - septum primum and septum secundum. If there is a deficit, perforation, or complete absence of septum secundum, this defect occurs. On the right atrial side, the defect is bounded by a protruding edge, known as limbus of fossa ovalis. The limbus septum separates the defect from the atrial walls, from the opening of the caval veins and the atrioventricular valves. Patent foramen ovale is not considered a severe pathology, it occurs in 25 - 30% of the population. It is not hemodynamically significant, but facilitates paradoxical embolization.
- Ostium primum ASD (type I defect).
It belongs to atrioventricular septal defects. At the same time, a cleft of the anterior mitral valve leaflet occurs and, unlike the complete form of atrioventricular septal defect, two separate rings of the atrioventricular valves are preserved. It accounts for 15-20% of defects of the interatrial septum.
- Sinus venosus superior ASD.
It is located beneath the inlet of the superior vena cava. The vein often saddles on the defect and opens up into both atria. It may be associated with anomalous return of the right pulmonary vein into the right atrium / superior vena cava. It accounts for about 10% of atrial septal defects.
- Sinus venosus inferior defect.
It is located at the inlet of the lower vena cava and is also associated with a partial anomalous return of the right pulmonary veins. The defect lacks a fixed lower edge. Eustachian valve may represent the false edge of the defect and derive blood from the lower vena cava into the left atrium. Thus, this type of defect may be an unknown cause of cyanosis in adulthood. It accounts for about 2% of defects.
- Coronary sinus defect (the unroofing of coronary sinus).
A defect of the interatrial septum at the coronary sinus inlet, associated with a different range of coronary sinus roof defect that separates it from the left atrium. It is associated with the persistence of the superior left vena cava, which flows into the left atrium or coronary sinus. It's very rare.

SYMPTOMS
If the defect is isolated, it is never critical. Children are asymptomatic and thrive normally, sometimes with more frequent respiratory infections. Often in adulthood, especially after the age of 40, exertional shortness of breath, fatigue (due to the development of pulmonary vascular obstructive disease and right heart failure), palpitations, recurrent respiratory infections occur. A serious complication is the paradoxical embolization of thrombi from the venous system through the atrial defect into the brain leading to stroke. Rarely, atrial flutter or paroxysmal supraventricular tachycardia occurs in children.
DIAGNOSIS
Auscultation: systolic murmur above the pulmonary artery, fixed split second heart sound above the pulmonary artery.
ECG: not a diagnostic method, an incomplete right bundle branch block and right axis deviation are common.
Chest X-ray: dilated pulmonary artery, dilated vessels of pulmonary hilus, increased pulmonary vascular congestion, raised heart apex as a result of dilatation of the right ventricle.
Echocardiography: the dominant diagnostic method. It can be used to evaluate the size and location of the ASD, the presence and severity of the interatrial shunt (including non-invasive Qp : Qs) and the degree of right ventricular overload.
Catheterization: is usually not necessary for diagnosis. It is performed in pulmonary hypertension to assess pulmonary vascular resistance and its reversibility, exceptionally when determining the hemodynamic significance of the defect or the course of anomalous pulmonary veins when it is unclear from a non-invasive examination.
OPERATION
Indication for surgery is given by severity of the left-to-right shunt (Qp: Qs) of 1.5: 1 and higher (that is a shunt greater than 30% of pulmonary flow). In girls it is sometimes recommended to close even smaller shunts because of the risk of paradoxical embolization in pregnancy. It is performed usually in preschool children, in some cases earlier. In the elderly, occlusion is indicated in case of (a) a flow rate greater than 2:1, (b) reversible pulmonary hypertension, or (c) venous thrombosis, i.e. history of stroke.
The principle of surgery is closing the defect by direct suture or patch (ideally by autologous pericardium) based on the size and shape of the defect, quality and compliance of the surrounding tissues - the suture must not be under tension due to the risk of complications. If there is an abnormal pulmonary vein return to the right atrium, it must be redirected back to the left atrium using the pericardial patch.
The standard surgical approach is lower partial sternotomy. An alternative approach is median sternotomy or short right-sided thoracotomy in the 5th intercostal space (cosmetic effect). Where robotic cardiac surgery is available, atrial septal defects are closed robotically. The surgery is performed with extracorporeal circulation in normothermia and with cardioplegic cardiac arrest, rarely in induced ventricular fibrillation.
Smaller ostium secundum ASD with well-formed edges can be closed by catheterization techniques.
RESULTS
The surgical risk (mortality) of operated children is less than 1% and the prognosis to adulthood is good. Possible (but rare) complications include residual shunt, bradyarrhythmias after injury of the heart conduction system, or postatriotomy tachyarrhythmias (atrial fibrillation or flutter).
Ventricular Septal Defect
 MORPHOLOGY
MORPHOLOGY
The defect represents a communication between the right and left ventricle. It is the most common congenital heart disease with a prevalence of 41.6%. It can be solitary, multiple, or in combination with other congenital heart defects. There are several anatomical variants depending on the location:
- Perimembranous defect
The most common (60-80% of all ventricular septal defects) is located in the upper, membranous, part of the ventricular septum just below the aortic valve (subaortic).
- Inlet defect
(8%) below the septal leaflet of the tricuspid valve, posteroinferior to the membranous septum.
- Outflow defect
(subarterial, subpulmonary, supracristal, infundibular; 5-7%). In Asians up to 30% of all ventricular septal defects, located above the supraventricular crest, beneath the semilunar valve.
- Muscular defect
(5-20%). It is surrounded by a muscular septum from all sides, localization can be central, apical or marginal, it can be multiple (sometimes referred to as ‘swiss cheese defect’).
Hemodynamically small defect is called restrictive, shunting is left-to-right and in the right ventricle retains significantly lower pressure than in the left ventricle. The non-restrictive defect is a large defect that allows free mixing of blood between the two chambers, the pressures in both chambers are balanced, shunting is bidirectional. Usually, a defect larger than the aortic orifice is considered a major defect.
In a ventricular defect, spontaneous closure of the pathological communication may occur, which occurs in 25 - 50% of cases within the first 3 years of life. The most common mechanism is the attachment of the hyperplastic septal leaflet of the tricuspid valve to the edges of the defect. Another way is the prolapse of the aortic valve leaflet (right or non-coronary) into the defect, but subsequently aortic regurgitation develops. Muscular defects may close by growth of the septum muscle.
Ventricular septal defect may also develop in adulthood, not as a congenital defect but as a mechanical complication of acute myocardial infarction. It leads to acute circulatory overload of the right ventricle leading to failure. It is a life-threatening condition with mortality above 80% within 4 weeks and requires urgent cardiac surgery.
SYMPTOMS
Small ventricular septal defects tend to be asymptomatic and manifest only as a murmur.
Medium and large defects lead to hyperdynamic pulmonary circulation, increased pulmonary congestion with airway obstruction and bronchial secretion. Tachypnoea, tiredness, dyspnoea and frequent respiratory infections occur. The volume-overload leads to the development of cardiomegaly with chest arching in the precordium.
Developed Eisenmenger syndrome (left-to-right shunt turns to right-to-left) manifests as cyanosis and exertional dyspnoea, but the condition may be stable for a relatively long time.
DIAGNOSIS
Auscultation: small and medium defects manifest as loud systolic murmur with a maximum in the left parasternal area, according to the localization of the defect in the 3rd - 5th intercostal space. A large defect causes no or only a quiet systolic murmur, the second heart sound above the pulmonary artery is accentuated.
ECG is not a diagnostic method, it may be quite normal, hypertrophy of both ventricles may be evident. With the development of pulmonary hypertension the heart axis deviates to the right.
Chest X-ray is not a diagnostic method, in a small defect it is normal, in a large defect there is dilation of the heart silhouette and an increased pulmonary vascular congestion.
Echocardiography: the main diagnostic method, it can determine the exact location of the defect, the size and the direction of shunting, estimation of pulmonary pressure, detection of associated defects.
Catheterization is performed to determine pulmonary vascular resistance and its reactivity to vasodilators. In the case where echocardiographic finding is unclear, it helps to quantify the size of the shunt.
OPERATION
Small and medium-sized defects with normal pulmonary vascular resistance tend to close spontaneously. They are operated only in case of complications (infectious endocarditis or aortic regurgitation).
Infants with a defect accompanied by uncontrollable heart failure require urgent surgical closure of the defect. In case of a complex heart defect or other paediatric problems, a pulmonary artery banding is performed, and a definitive solution of the defect is delayed.
For children who have no significant difficulties and have left-to-right shunt Qp / Qs of 2: 1 and greater, it is best to operate between the ages of 2-3 years. In older patients, the indication for closure is the same. Contraindications are pulmonary vascular resistance greater than 10 Wood units. In borderline cases, tests to reduce pulmonary vascular resistance and / or pulmonary biopsy are a key to making a decision.
The operation is performed with extracorporeal circulation with cardioplegic cardiac arrest through median sternotomy. The defect is usually closed by sewing in a patch from artificial material. Depending on the location, access to ventricular defects is achieved through the right atrium and tricuspid valve, through the outflow tract of the right ventricle, and rarely in apically localized defects through left ventriculotomy.
Palliative banding of the pulmonary artery is performed through a left-sided thoracotomy. First, the ductus arteriosus is ligated. A silastic strip is passed around the pulmonary trunk and is being tightened until the pulmonary artery pressure drops to 1/3 of the system pressure. At the same time, peripheral blood saturation must not fall below 90%.
Recently, percutaneous occlusion has been increasingly performed with special occluders.
RESULTS
The surgical risk of planned operations of isolated ventricular defects without or with only low pulmonary hypertension is low and the long-term results are excellent. Surgical risk is increased in critical defects, in infants and in patients with high pulmonary hypertension.
Rare but possible postoperative complications include: recanalization defect, AV-block, tricuspid valve regurgitation, infectious endocarditis, ventricular dysrrhythmias.
Tetralogy of Fallot
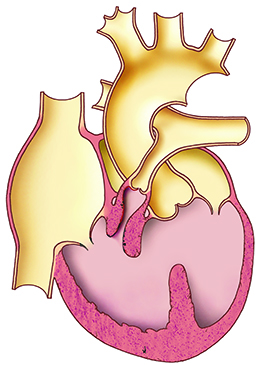 MORPHOLOGY
MORPHOLOGY
Complex congenital heart defect defined by a tetrad of:
- large non-restrictive ventricular septal defect (subaortic)
- an overriding aorta above this defect ("aortic dextroposition")
- right ventricular outflow tract obstruction caused by infundibular stenosis of the pulmonary artery, valvular stenosis of the pulmonary artery, or possibly supravalvular stenosis of the pulmonary artery
- secondary right ventricular hypertrophy
It was clinically defined by Arthur Fallot in 1888. Also described is Fallot's pentalogy, in which the aforementioned tetrad of symptoms is extended by an atrial septal defect.
SYMPTOMS
The essential manifestation is central cyanosis due to obstruction of the right ventricular outflow tract. It is a result of expelling deoxygenated blood from the right ventricle through the large subaortic ventricular septal defect into the overriding aorta, only a small portion passes through the valvular stenosis into the pulmonary artery. In new-borns, cyanosis can dramatically worsen after the spontaneous occlusion of the ductus arteriosus, after which pulmonary flow is significantly reduced. Tet spells (acute hypoxia spells) with severe cyanosis and hyperpnoea occur in unoperated children with protracted desaturation due to increased contractions of the right ventricular infundibulum. In these tet spells, children squat, increasing the venous return and systemic vascular resistance and subsequently reducing the right-to-left shunt. Pharmacologically, the spell can be suppressed by administration of beta-blockers on the principle of reducing myocardial contractility. Chronic cyanosis leads to secondary erythrocytosis (polyglobulia). Typical symptoms are digital clubbing (nails are shaped like an hourglass). Rarely, there are patients with mild forms of Fallot's tetralogy with mild pulmonary obstruction and low overriding aorta, who may be almost cyanosis-free and can live to adulthood without surgery and without major discomfort (the so-called "pink tet" or pink tetralogy of Fallot).
DIAGNOSIS
Auscultation: a loud systolic murmur of crescendo-decrescendo character with a maximum in the 3rd intercostal space parasternally on the left (above the right ventricle infundibule).
Aspect: central cyanosis, digital clubbing.
ECG: isolated right ventricular hypertrophy.
Chest X-ray: The raised apex in right ventricular hypertrophy and concavity at the pulmonary site (pulmonary hypoplasia) gives the heart a typical shape ("coeur en sabot" or "boot shaped"). Poor pulmonary vascular filling due to decreased pulmonary flow.
Echocardiography: a dominant diagnostic method. In addition to diagnosis, it exludes other associated defects and allows differential diagnosis of cyanotic heart defects.
Catheterization: its importance decreased with the development of echocardiography. Catheterization nowadays specifies the morphology of the outflow tract of the right ventricle, pulmonary artery and its branches, morphology and coronary artery branching, it displays aortopulmonary collaterals and hemodynamic data which are important before surgery. It helps to better visualize the morphology of large vessels after previous shunting operations.
OPERATION
Infants with cyanosis and severe tet spells not responding to pharmacotherapy are indicated for surgery due to defect severity. If they have a well-developed pulmonary vascular system and do not have a hypoplastic left ventricle, we perform a radical correction of the defect at 6-12 months. In pulmonary arterial or left ventricular hypoplasia, an arterial shunting operation is first performed. As soon as the increase in pulmonary circulation improves the capacity of the pulmonary vascular system and the left ventricle, a definitive correction of the defect can be performed in the second stage.
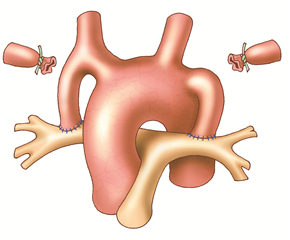
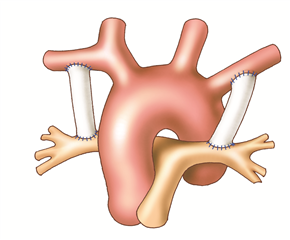 The principle of palliative shunting operations is to bring more mixed blood to the lungs from the systemic circulation. The best known is the Blalock-Taussig shunt (subclaviopulmonary anastomosis) where the end of the subclavian artery is anastomosed to the side of the pulmonary artery. Nowadays, the so-called modified Blalock shunt is used more frequently, where the connection between the subclavian artery and the pulmonary artery is performed using an interposed goretex prosthesis. It is done through right- or left-sided thoracotomy depending on the choice of subclavian artery (right is preferred). Another possibility is to create a so-called central shunt seu Amato (anastomosis of the ascending aorta and pulmonary trunk with a prosthesis) through left-sided thoracotomy or median sternotomy.
The principle of palliative shunting operations is to bring more mixed blood to the lungs from the systemic circulation. The best known is the Blalock-Taussig shunt (subclaviopulmonary anastomosis) where the end of the subclavian artery is anastomosed to the side of the pulmonary artery. Nowadays, the so-called modified Blalock shunt is used more frequently, where the connection between the subclavian artery and the pulmonary artery is performed using an interposed goretex prosthesis. It is done through right- or left-sided thoracotomy depending on the choice of subclavian artery (right is preferred). Another possibility is to create a so-called central shunt seu Amato (anastomosis of the ascending aorta and pulmonary trunk with a prosthesis) through left-sided thoracotomy or median sternotomy.
The radical operation of Fallot's tetralogy consists in closure of the subaortic ventricular septal defect with a patch, resolution of pulmonary stenosis and widening of the narrowed right ventricualr outflow tract. It is performed with extracorporeal circulation with cardioplegic cardiac arrest through a median sternotomy approach. Intracardial surgery is performed through the right atrium and pulmonary trunk (transatrial-transpulmonary approach). It replaced the original transventricular approach, which increased the risk of arrhythmias and right ventricular dysfunction in the long-term postoperatively. When intervention on the hypoplastic pulmonary valve annulus is required, pericardial patch is used for widening, sometimes with an artificially created pericardial valve. However, post-operative pulmonary regurgitation of various severity occurs. Mild is not prognostically serious, but significant regurgitation leads to failure of the right ventricle after years due to volume overload. In these rare cases, the condition should be resolved by implanting preferably a biological valve into the pulmonary orifice.
RESULTS
Operational risk is currently below 1%. The prognosis after radical surgical correction of Fallot's tetralogy is good. Outpatient cardiologist monitoring is necessary due to various complications: recanalization of the defect, residual obstruction of the right ventricular outflow tract, pulmonary valve or pulmonary artery, pulmonary regurgitation and right heart failure, dysrrhythmias (from the scar area after ventriculotomy), right ventricular aneurysm near ventriculotomy, infectious endocarditis.
Transposition of the Great Arteries
 MORPHOLOGY
MORPHOLOGY
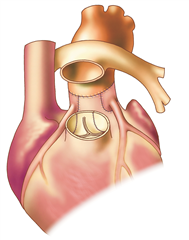
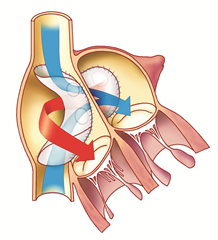
This anomaly is considered to be uncorrected transposition of the great arteries. The heart cavities are arranged in atrioventricular concordance and the offsets of the great arteries are in ventriculoarterial discordance (the large arteries therefore exit from the opposite ventricles - the aorta from the right ventricle and the pulmonary artery from the left ventricle). About one half to two thirds of patients have isolated transposition, without associated defects, the remaining defects are complex, most often associated with ventricular septal defect or obstruction of the outflow tract of the left ventricle (subpulmonary stenosis). Uncorrected transposition of great arteries stands for two independent blood circulations that are incompatible with life when there is no communication between them. After birth, the communication (shunting) is ensured for several days by an open foramen ovale and a patent ductus arteriosus (and possibly a ventricular septal defect). Once the natural shunt closes, the defect quickly leads to critical hypoxemia and death of the infant.
The right ventricular hypertrophy adapts to systemic pressure if the shunts are preserved. The left ventricular myocardium, on the other hand, remains weak due to the low pulmonary resistance.
SYMPTOMS
Symptoms appear right after birth and depend on the specific morphology of the defect. The defect is always critical. Cyanosis, tachypnoea, dyspnoea, tachycardia and congestive heart failure occur.
DIAGNOSIS
Auscultation findings are poor.
ECG: signs of right ventricular hypertrophy.
Chest X-ray shows apparently enlarged egg-shaped heart with narrow vessel silhouette and increased pulmonary vascular congestion.
Echocardiography: main diagnostic method. It shows the morphology of the defect, branching of the great arteries and coronary arteries from the aorta, size and shape of the ventricles, and other associated anomalies.
Catheterization is not necessary for diagnosis, but allows for life-saving intervention - balloon atrioseptostomy.
OPERATION
In isolated transposition, urgent balloon atrioseptostomy, according to Rashkind, and administration of prostaglandin E1 - to maintain natural circulatory shunts (foramen ovale and ductus arteriosus) are essential to perform after birth. Anatomical correction of the defect sec Jatene(so called “arterial switch operation”) must be performed within 2 weeks, 3 weeks at the latest, before the left ventricle becomes less adapted to pressure load. In infants who have not undergone early anatomical correction in the neonatal age, the left ventricle must be "trained" prior to anatomic correction by performing a pulmonary artery banding and a shunting operation (a vascular prosthesis connects the left subclavian artery with the left pulmonary artery). After 7-10 days, this pressure-acclimatized left ventricle can be connected to the systemic circulation using Jatene procedure. In the case of a late-diagnosed transposition, training can be performed later, but requires a longer period of acclimatization (up to 1 year).
Anatomical correction of the defect (Jatene procedure, arterial switch) is a radical procedure at the level of the large arteries, where the aorta and pulmonary artery are resected and exchanged, and the coronary arteries are reimplanted into the neo-aorta. Preferred procedure.
Physiological corrections of the defect (Mustard procedure or Senning procedure) are procedures at the atrial level, nowadays they are no longer performed for uncorrected transposition. The principle is to direct all the venous blood flowing into the right atrium from both the superior and inferior vena cava into the left ventricle from which the pulmonary artery arises. Blood from the pulmonary veins is completely derived via the left atrium to the right ventricle and to the aorta. This can be achieved by constructing a tunnel inside the atrium with pericardium (Mustard operation) or by using the right atrial wall (Senning operation).
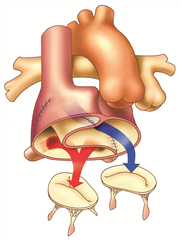
RESULTS
Operational mortality of physiological correction is around 3% and of anatomical correction between 1-2%. Without surgery, 90% of children die within the first year of life. Operated infants have good life prognosis. In adulthood, however, after physiological corrections, right (systemic) ventricular dysfunction with significant tricuspid regurgitation may develop, as well as dysrrhythmia, stenosis of the superior or inferior vena cava, and pulmonary vein obstruction. After anatomical correction, possible complications include myocardial ischemia from kinks or stenosis of the orifice of the implanted coronary arteries and also the development of aortic valve regrugitation.
Resources:
- Hučín B. Dětská kardiochirurgie - 2., doplněné vydání. Praha: Grada, 2012. ISBN 978-80-247-4497-1.
- Češka R a kol. Interna. Praha: Triton, 2010. ISBN: 978-80-7387-423-0.
- Dominik J. Kardiochirurgie. Praha: Grada Publishing, 1998. ISBN: 80-7169-669-2.
- Dominik J.: Kardiochirurgie. In.: Pafko P. et al.: Základy speciální chirurgie, Galén, 2008, s.173-195. ISBN 978-80-7262-402-7
We hereby thank Roman Gebauer, M.D., chief physician of the Cardiac Surgery Department at Paediatric cardio center in Motol, Prague, for the consultation and actualization of the data.
