Vrozené vady
Otevřená tepenná dučej
 MORFOLOGIE
MORFOLOGIE
Otevřená tepenná dučej spojuje ve fetální cirkulaci plicnici se sestupnou aortou, do níž ústí z mediální strany, distálně od odstupu levé podklíčkové tepny. Svalová tkáň dučeje je citlivá na prostaglandin placenty v průběhu gravidity. Po porodu dochází k poklesu hladiny prostaglandinu v krvi, co má za následek kontrakci svaloviny a uzávěr dučeje. U donošeného novorozence se uzavírá do několika dnů až do 3 týdnů. Stěna perzistující otevřené dučeje má jinou histologickou strukturu – chybí u ní typická kontraktilní schopnost.
Zkrat dučejí je podle tlakového spádu levo-pravý, tedy z aorty do plicnice a krev proudí tímto směrem nejen v systole, ale i v diastole. Plicní průtok je zvýšen. Velký levo-pravý zkrat zvyšuje tlak v plicním cévním řečišti a podporuje rozvoj plicní cévní obstrukční choroby. Postupně vzniká těžká ireverzibilní plicní hypertenze a může dojít až k obrácení toku ve zkratu na pravo-levý (tzv. Eisenmengerův syndrom). Tento tok již není typicky kontinuální.
SYMPTOMY
Malá dučej může být klinicky němá až do dospělosti, projevuje se pouze šelestem. U velké dučeje vzrůstá levo-pravý zkrat při poklesu plicní rezistence po narození. Zvýšený plicní průtok způsobuje objemové přetížení levé komory, dochází k srdečnímu selhávání až plicnímu edému. Projevuje se dyspnoí, tachypnoí a častými respiračním infekty.
Při vysoké plicní rezistenci u Eisenmengerova syndromu je u otevřené tepenné dučeje cyanóza pouze v dolní polovině těla (pod ústím tepenné dučeje do aorty), terminálně může dojít i k pravostranné kardiální dekompenzaci.
DIAGNÓZA
Poslechově je přítomen typický kontinuální (systolicko-diastolický) šelest pod levým klíčkem. U Eisenmengerova syndromu u otevřené tepenné dučeje je pouze systolický šelest, nebo akcentace 2. ozvy nad arteria pulmonalis.
EKG bývá normální, se zvyšující se hemodynamickou závažností zkratu se mohou objevit známky objemového zatížení levé komory.
Na RTG je zvětšený srdeční stín a bohatá plicní vaskulární kresba.
Echokardiografie je pro určení diagnózy nejdůležitější. Pomocí dvourozměrné a dopplerovské echokardiogradiografie jsme schopni určit velikost, umístění i hemodynamickou významnost zkratu.
Katetrizace je prováděna u Eisenmengerova syndromu k určení reverzibility plicní hypertenze. Díky včasné diagnostice a operaci však již v současnosti Eisenmengerův syndrom u otevřené tepenné dučeje nevídáme.
OPERACE
Indikace k uzávěru otevřené tepenné dučeje. U nedonošených symptomatických novorozenců je indikován pokus o farmakologický uzávěr zkratu podáním indometacinu nebo ibuprofenu (antagonisty syntézy prostaglandinu). Při neúspěchu se přistupuje k operaci. U novorozenců a kojenců se vada operuje pouze při nezvládnutelné srdeční slabosti (kritická vrozená srdeční vada). U asymptomatických dučejí je vhodné uskutečnit operaci plánovaně v batolecím věku.
Operace je kontraindikována při rozvinutém Eisenmengerovém syndromu nebo v přítomnosti komplexních vad, kde je cirkulace krve závislá na perzistenci zkratu (atrézie plicnice, Fallotova tetralogie, nekorigovaná transpozice velkých tepen atd.). V dospělosti je operace zcela výjimečná a většinou je spojena s velkým rizikem pro již velmi křehkou a kalcifikovanou stěnu dučeje i pro již přítomnou plicní hypertenzi.
První úspěšný operační uzávěr tepenné dučeje v České republice byl proveden v roce 1947 prof. Bedrnou v Hradci Králové.
Operačním přístupem je levostranná torakotomie. Po vypreparování dučeje je tato uzavřena pomocí 2 - 3 ligatur. Alternativní metodou je resekce dučeje s prošitím obou pahýlů, což je nezbytné u široké a současně krátké dučeje – dnes je prováděna již jen vzácně. Uzavřít otevřenou tepennou dučej je dnes možné i torakoskopicky (naložení klipu) nebo katetrizační technikou (pomocí spirály, která vyplní lumen celé dučeje).
VÝSLEDKY
Operační riziko uzávěru izolované tepenné dučeje při plánovaných operacích u malých dětí je nižší než 1 %. Zvyšuje se u nedonošených dětí, u kritických vad a také u vzácných operací v dospělosti.
Prognóza po úspěšném uzávěru dučeje je dobrá. Pokud nedošlo k uzávěru hemodynamicky nevýznamné dučeje, prognóza do dospělosti je dobrá, ale nemocní jsou ohroženi vývojem endarteritidy v dučeji. Významnější zkrat je v dospělosti vzácný. Je řešitelný standardními způsoby, pokud se ale jedná už o rozvinutý Eisenmengerův syndrom, prognóza je špatná.
Koarktace aorty
 MORFOLOGIE
MORFOLOGIE
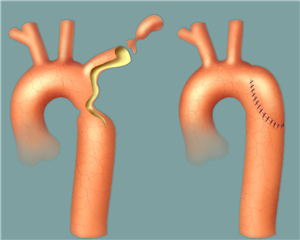
Koarktace je zúžení aorty lokalizované většinou těsně pod odstupem levostranné podklíčkové tepny v oblasti vústění tepenné dučeje, v oblasti tzv. isthmus aortae. Je způsobená vnitřní lištou, vyklenující se ze stěny aorty proti ústí tepenné dučeje. Zúžení průsvitu aorty novorozence pod 2 mm se označuje za těsné, poněvadž představuje významnou překážku toku krve v descendentní aortě, která je široká 5 mm. Pod místem koarktace bývá aorta poststenoticky dilatována.
Méně často je koarktace provázena i hypoplazií aortálního oblouku a/nebo delším tubulárním zúžením istmu. Extrémní varianty představují interrupci (přerušení) nebo atrézii aortálního oblouku (celý oblouk je nahrazen vazivovým pruhem). Koarktace aorty je často sdružena s bikuspidální chlopní aorty; současně mohou být přítomny i jiné vrozené srdeční vady, např. defekt komorového septa.
SYMPTOMY
Nejzákladnějším projevem koarktace aorty je systémová hypertenze v horní polovině těla s tlakovým přetížením levé komory. Přes a. thoracica interna a interkostální tepny se vytváří kolaterální oběh do descendentní aorty, který zabezpečuje prokrvení dolní poloviny těla. V případě, že není vytvořen (zejména u novorozenců), projevuje se koarktace jako kritická vada s levostranným srdečním selháním a cirkulačním kolapsem. Po přirozeném uzávěru tepenné dučeje dochází k hypoperfuzi postkoarktační oblasti se selháním ledvin, oligurií, metabolickou acidózou a multiorgánovým selháním.
Koarktace aorty s dobře vytvořeným kolaterálním oběhem se projeví až v pozdějším dětském nebo dospělém věku. Je přítomna hypertenze na horních končetinách, cefalea, epistaxe, slabost a únavnost dolních končetin, může být přítomna námahová dušnost a bolesti na hrudi. K závažným komplikacím patří mozkové krvácení a disekce nebo ruptura aorty.
DIAGNÓZA
Poslech: systolický šelest nad aortou a mezi lopatkami, kolaterály se projeví kontinuálním šelestem v mezižeberních prostorech.
Palpačně: vymizelá nebo výrazně oslabená a opožděná pulzace na femorálních tepnách. Krevní tlak je nižší na dolních končetinách oproti horním končetinám. Za významný a pro koarktaci svědčící je považován tlakový gradient nad 20 mm Hg.
EKG: hypertrofie levé komory. Při současném "přetížení" s depresemi ST-úseků může být koarktace sdružena s významnou aortální stenózou.
RTG srdce a plic: zářez na sestupné hrudní aortě v místě koarktace (znamení trojky – krátké ligamentum arteriosum deformuje aortální isthmus do tvaru čísla „3“), usurace spodních částí žeber širokými kolaterálami interkostálních tepen. Může, ale nemusí být přítomna kardiomegalie a městnání v malém oběhu.
Echokardiografie: suprasternálně dvourozměrnou echokardiografií hodnotíme morfologii aortálního oblouku a odstup velkých cév, dopplerovsky lze odvodit gradient na koarktaci měřením zvýšené systolické rychlosti v sestupné hrudní aortě, typické je současné diastolické dopředné proudění v místě koarktace.
Katetrizace a aortografie: určení gradientu na koarktaci, přesné zhodnocení morfologie koarktace, přítomnost kolaterál, aneuryzmat, abnormalit v odstupu velkých tepen, koronarografie.
OPERACE
Indikace k operaci je urgentní u novorozenců a kojenců v případě nezvládnutelné levostranné srdeční slabosti. Elektivní operaci je nejlépe uskutečnit ve věku 3 - 5 let. Provádí se především u asymptomatických dětí se systémovou hypertenzí. U dětí bez hypertenze je indikována při významném vzestupu gradientu na koarktaci po zátěží (z 10 na 60 mmHg), pří významné hypertenzní reakci na zátěž, při ischémii myokardu, nebo při závažné deformaci aorty v místě isthmu (tvar čísla „3“).
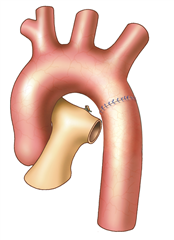
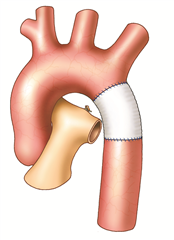
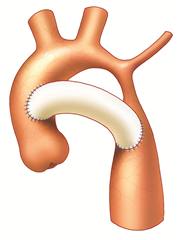
Operace koarktace je prováděna z levostranné torakotomie. Dle morfologického nálezu je možné zvolit jednu z mnoha technických variant řešení. Nejčastěji a nejsprávněji je to kompletní resekce koarktace a anastomóza end-to-end. V případě delšího zúženého úseku, je používána plastika koarktace dle Waldhausena, při které se oblast koarktace podélně protne a rozšíří lalokovou plastikou z podvázané a sklopené levé podklíčkové tepny. Další možností je rozšíření koarktace všitím záplaty dle Vosschulteho. V případě dlouhého patologicky změněného úseku aorty (hypoplazie, aneurysma) je nutno tento resekovat a uskutečnit náhradu cévní protézou. U rekoarktací a dospělých jsou někdy využívány bypassové operace (vytvoření aortoaortálního nebo subklavioaortálního bypassu). V úvahu přichází i řešení metodami intervenční kardiologie (dilatace, stentování).


VÝSLEDKY
Operační mortalita je nízká. Peroperační a pooperační komplikace jsou sice vzácné, ale velmi závažné (krvácení ze sutury nebo křehkých kolaterál, paradoxní pooperační hypertenze s parézou střev, ischémie míchy s paraplegií dolních končetin, pseudoaneurysma v okolí cévní protézy, přetrvávání hypertenze u pozdě operovaných). U některých operovaných se může rozvinout rekoarktace (udává se riziko do 20%). Pokud se jedná o hemodynamicky významnou rekoarktaci, řeší se v současnosti pomocí balonkové perkutánní angioplastiky, eventuálně v kombinaci s implantací kovového stentu. Při neúspěchu této metody přistupujeme k reoperaci.
Defekt síňového septa
 MORFOLOGIE
MORFOLOGIE
Defekt síňového septa znamená patologickou komunikaci mezi pravou a levou síní. Defekt umožňuje patologický tok krve mezi síněmi. Směr zkratu je levo-pravý z důvodu větší poddajností pravé síně a pravé komory. Dochází ke zvýšenému plicnímu průtoku, dilataci a objemovému přetížení pravé komory a plicnice, při trikuspidální regurgitaci i pravé síně. Levá komora zůstává malá. Poměr zvýšeného průtoku krve plícemi k systémovému průtoku (Qp : Qs) je kvantifikací zkratu. Síňové defekty se zpravidla spontánně neuzavírají.
Defekt může být umístěn v různých oblastech mezisíňové přepážky. Umístění defektu je podmíněno způsobem jeho embryonálního vzniku a ovlivňuje techniku chirurgického uzávěru.
- Defekt síňového septa typu ostium secundum (defekt II. typu, defekt ve fossa ovalis)
Nejčastější typ (asi 70 % všech defektů na úrovni síní). Foramen ovale, které tvoří důležitou strukturu pro pravo-levý zkrat ve fetální cirkulaci, se uzavírá v prvním týdnu po narození slepením obou řas, které jej tvoří – septum primum a septum secundum. Při deficitu, perforaci, nebo úplném chybění septum secundum vzniká tento defekt. Ze strany pravé síně je defekt ohraničen vystouplým okrajem, tzv. limbus fossae ovalis. Limbické septum odděluje defekt od stěn síně, ústí dutých žil a atrioventrikulárních chlopní. Otevřené foramen ovale není považováno za zásadní patologii, vyskytuje se u 25 - 30 % populace. Hemodynamicky se neuplatňuje, ale umožňuje paradoxní embolizaci.
- Defekt síňového septa typu ostium primum (defekt I. typu)
Řadí se mezi defekty atrioventrikulárního septa. Současně s ním se vyskytuje i rozštěp předního cípu mitrální chlopně a na rozdíl od úplné formy defektu atrioventrikulárního septa ještě jsou zachovány dva oddělené prstence atrioventrikulárních chlopní. Tvoří 15-20% defektů mezisíňové přepážky.
- Defekt typu sinus venosus superior
Nachází se pod vústěním horní duté žíly, která na něj často nasedá a ústí tak do obou síní. Bývá spojen s anomálním návratem pravostranní horní plicní žíly do pravé síně, resp. až do horní duté žíly. Tvoří asi 10% síňových defektů.
- Defekt typu sinus venosus inferior
Je lokalizován v místě vústění dolní duté žíly a bývá též spojen s částečným anomálním návratem pravostranných plicních žil. U defektu chybí pevný dolní okraj. Eustachova řasa může představovat falešný okraj defektu a derivovat krev z dolní duté žíly do levé síně. Tento typ defektu tak může být nepoznanou příčinou cyanózy v dospělosti. Tvoří asi 2% defektů.
- Defekt koronárního sinu (nezastřešený koronární sinus, „unroofing“)
Defekt mezisíňové přepážky v místě vústění koronárního sinu, spojený s různě rozsáhlým defektem stropu koronárního sinu, který jej odděluje od levé síně. Bývá spojen s perzistencí levostranné horní duté žíly, která ústí do levé síně nebo do koronárního sinu. Je velmi vzácný.

SYMPTOMY
Pokud je vada izolovaná, není nikdy kritická. Děti bývají asymptomatické a normálně prospívají, někdy se u nich objevují častější respirační infekty. Často až v dospělosti, především po 40. roce věku, se objevuje námahová dušnost, únavnost (z důvodu rozvoje plicní cévní obstrukční choroby a pravostranného kardiálního selhání), palpitace, recidivující respirační infekty. Závažnou komplikací je paradoxní embolizace trombů ze žilního řečiště síňovým defektem do mozku se vznikem cévní mozkové příhody. Vzácně se i u dětí objevuje flutter síní nebo paroxyzmální supraventrikulární tachykardie.
DIAGNÓZA
Poslech: systolický šelest nad plicnicí, fixní rozštěp 2. ozvy nad plicnicí.
EKG: nepatří mezi diagnostické metody, častý je inkompletní blok pravého Tawarova raménka a sklon srdeční osy doprava.
RTG srdce a plic: rozšířená plicnice, rozšířené cévy v plicních hilech, zvýšená plicní cévní kresba, nadzdvižený srdeční hrot při dilataci pravé komory.
Echokardiografie: hlavní diagnostická metoda. Je schopná popsat velikost a uložení síňového defektu, přítomnost a závažnost zkratu v síních (včetně neinvazivního určení poměru Qp : Qs) i stupeň zátěže pravé komory.
Katetrizace: pro diagnózu není většinou nutná. Provádí se u plicní hypertenze ke zhodnocení výše plicní vaskulární rezistence a její reverzibility, výjimečně při nutnosti stanovení hemodynamické významnosti defektu nebo průběhu anomálních plicních žil, není-li toto jasné z neinvazivního vyšetření.
OPERACE
Indikace k operaci je dána levopravým zkratem (Qp : Qs) 1,5 : 1 a vyšším (tj. zkrat větší než 30 % plicního průtoku). U dívek je někdy doporučováno uzavírat i menší zkraty pro riziko paradoxní embolizace v těhotenství. Provádí se standardně u dětí předškolního věku, výjimečně dříve. U starších pacientů je uzávěr indikován v případě (a) poměru průtoků většího než 2:1, (b) plicní hypertenze, která je ještě reverzibilní, nebo (c) žilní trombózy, ev. cévní mozkovou příhodu v anamnéze.
Principem chirurgického řešení je uzávěr defektu přímou suturou nebo záplatou (ideálně autologním perikardem) na základě velikosti a tvaru defektu, kvality a poddajnosti okolních tkání – sutura nesmí být pod tahem z důvodu rizika komplikací. Pokud je přítomen abnormální návrat plicních žil do pravé síně, je nutno je perikardiální záplatou derivovat zpět doleva.
Standardním operačním přístupem je dolní parciální sternotomie. Alternativním přístupem je podélná sternotomie nebo krátká pravostranná torakotomie v 5. mezižebří (kosmetický efekt). Na pracovištích, kde je zavedena robotická kardiochirurgie, se defekty síňového septa uzavírají roboticky. Operace se provádí v mimotělním oběhu v normotermii a kardioplegické srdeční zástavě, vzácněji v navozené fibrilaci komor.
Menší defekty síňového septa typu ostium secundum s dobře vytvořenými okraji lze uzavřít katetrizační technikou.
VÝSLEDKY
Operační riziko (mortalita) je u operovaných dětí nižší než 1 % a prognóza do dospělosti je dobrá. Mezi možné (ale vzácné) komplikace řadíme reziduální síňový defekt, bradyarytmie po poranění převodního systému, nebo postatriotomické tachyarytmie (fibrilace nebo flutter síní).
Defekt komorového septa
 MORFOLOGIE
MORFOLOGIE
Defekt představuje komunikaci mezi pravou a levou komorou. Je vůbec nejčastější vrozenou srdeční vadou s prevalencí 41,6 %. Může být solitární, mnohočetný, nebo jako kombinace s jinými vrozenými srdečními vadami. Existuje několik anatomických variant v závislosti od lokalizace:
- Perimembranózní defekt
Nejčastější (60 - 80 % všech defektů komorového septa), je lokalizován v horní membranózní části komorového septa těsně pod aortální chlopní (subaortální).
- Vtokový defekt
(8 %) pod septálním cípem trikuspidální chlopně, posteroinferiorně od membranózního septa.
- Výtokový defekt
(subarteriální, subpulmonální, suprakristální, infundibulární; 5 - 7 %). U Asiatů až 30 % všech defektů komorového septa, lokalizován nad crista supraventricularis, pod semilunárními chlopněmi.
- Muskulární defekt
(5 - 20 %). Ze všech stran ohraničen muskulárním septem, lokalizace může být centrální, apikální nebo marginální, může být i mnohočetný (někdy vytváří až obraz ementálského sýra, tzv. „swiss cheese defect“).
Hemodynamicky malý defekt označujeme jako restriktivní, zkrat je levo-pravý a v pravé komoře zůstává významně nižší tlak než v levé komoře. Nerestriktivní defekt je velký defekt, který umožňuje volné mísení krve mezi oběma komorami, tlaky v obou komorách jsou vyrovnané, zkrat je obousměrný. Obyčejně je za velký defekt považován defekt větší jako aortální ústí.
U komorového defektu může dojít ke spontánnímu uzávěru patologické komunikace, děje se tak ve 25 - 50 % případů v průběhu prvních 3 let života. Nejčastějším mechanizmem je přiložení hyperplastického septálního cípu trikuspidální chlopně k okrajům defektu. Jinou variantou je prolaps cípu aortální chlopně (pravého nebo nekoronárního) do defektu, následně se ale rozvíjí aortální regurgitace. Muskulární defekty se uzavírají narůstáním svaloviny septa.
Defekt komorového septa se může rozvinout taky v dospělosti, nikoliv jako vrozená vada, ale jako mechanická komplikace akutního infarktu myokardu. Vede k akutnímu oběhovému přetížení pravé komory s jeho selháním. Jedná se o život ohrožující stav s mortalitou nad 80 % do 4 týdnů a vyžaduje časný kardiochirurgický výkon.
PŘÍZNAKY
Malý defekt komorového septa bývá asymptomatický a projeví se pouze šelestem.
Středně velký a velký defekt vede k hyperkinetické plicní cirkulaci a plicnímu městnání s obstrukcí dýchacích cest a zvýšenou bronchiální sekrecí. Projeví se tachypnoí, únavností, dušností a častými respiračními infekty. Objemová zátěž vede k rozvoji kardiomegalie s vyklenováním hrudníku v prekordiu.
Rozvinutý Eisenmengerův syndrom (levo-pravý zkrat se změní na pravo-levý) se projeví cyanózou a námahovou dušností, stav však může být relativně dlouho stabilizován.
DIAGNÓZA
Poslechově se malý a střední defekt projeví hlučným systolickým šelestem s maximem vlevo parasternálně, podle lokalizace defektu ve 3. - 5. mezižebří. Velký defekt nezpůsobuje žádný, nebo jenom malý systolický šelest, je akcentovaná druhá ozva nad plicnicí.
EKG není diagnostickou metodou, může být zcela normální, může být patrná hypertrofie obou komor, s rozvojem plicní hypertenze se srdeční osa stáčí doprava.
RTG srdce a plic není diagnostickou metodou, u malého defektu je normální, u velkého dilatace srdečního stínu a zmnožená plicní cévní kresba.
Echokardiografie: hlavní diagnostická metoda, umožňuje určit přesnou lokalizaci defektu, velikost a směr zkratu, odhad tlaku v plicnici, detekce přidružených vad.
Katetrizace se provádí ke zjištění plicní cévní rezistence a její reaktivity na vazodilatační látky, případně při nejasném echokardiografickém nálezu pomáhá kvantifikovat velikost zkratu.
OPERACE
Malé a středně velké defekty s normální plicní cévní rezistencí mají tendenci ke spontánnímu uzávěru. Operují se jenom v případě komplikací (infekční endokarditida nebo aortální regurgitace).
Kojenci s defektem provázeným nezvládnutelnou srdeční slabostí vyžadují urgentní operační uzávěr defektu. V případě komplexní srdeční vady nebo nasedajících jiných pediatrických problémů se provádí bandáž plicnice a definitivní řešení vady přichází na řadu až v druhé době.
U dětí, které nemají výrazné potíže a mají levo-pravý zkrat Qp/Qs 2 : 1 a větší, je nejlépe operovat ve věku 2 - 3 let. U starších nemocných je indikace k uzávěru stejná. Kontraindikací je plicní vaskulární rezistence větší než 10 Woodových jednotek. V hraničních případech rozhodnou testy na snížení plicní vaskulární rezistence, případně plicní biopsie.
Operaci provádíme v mimotělním oběhu s kardioplegickou srdeční zástavou přístupem z mediánní sternotomie. Defekt se zpravidla uzavírá všitím záplaty z umělé hmoty. Vlastní přístup ke komorovým defektům je dle jejich lokality volen přes pravou síň a trikuspidální chlopeň, přes výtokový trakt pravé komory, zřídka u apikálně lokalizovaných defektů možno využít i ventrikulotomii levé komory.
Paliativní bandáž plicnice se provádí z levostranné torakotomie. Nejprve se podváže tepenná dučej. Kolem kmene plicnice se provlékne silastikový proužek, který se stahuje až do momentu, kdy tlak v plicnici poklesne na 1/3 systémového tlaku. Saturace periferní krve současně nesmí poklesnout pod 90 %.
V poslední době se stále častěji zkouší i perkutánní uzávěr speciálním okludérem.
VÝSLEDKY
Operační riziko plánovaných operací izolovaných komorových defektů bez anebo s nízkou plicní hypertenzí je malé a dlouhodobé výsledky jsou výborné. Operační riziko se zvyšuje u kritických vad, u kojenců a u operovaných s vysokou plicní hypertenzí.
K vzácným, ale možným pooperačním komplikacím patří: rekanalizace defektu, AV-blokáda, insuficience trikuspidální chlopně, infekční endokarditida, komorové dysrytmie.
Fallotova tetralogie
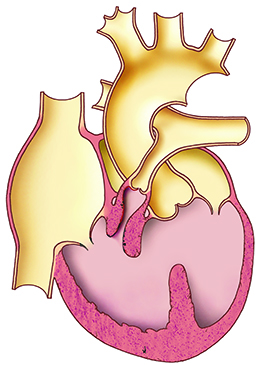
MORFOLOGIE
Komplexní vrozená srdeční vada definovaná tetrádou:
- velký nerestriktivní defekt komorového septa (subaortálně uložený)
- na tento defekt nasedající aorta („dextropozice aorty“)
- obstrukce výtokového traktu pravé komory způsobená infundibulární stenózou plicnice, valvární stenózou plicnice, případně i supravalvární stenózou plicnice
- sekundární hypertrofie pravé komory
Klinicky byla definovaná Arthurem Fallotem v roce 1888. Popisována je i Fallotova pentalogie, u které je tetráda příznaků rozšířena ještě o defekt síňové přepážky
SYMPTOMY
Základním projevem je centrální cyanóza z důvodu obstrukce výtokového traktu pravé komory. Vzniká tím, že pravá komora vypuzuje žilní krev převážně velkým subaortálním defektem do nasedající aorty, jen malou část přes stenózu do plicnice. U novorozenců se může cyanóza dramaticky prohloubit po spontánním uzávěru otevřené tepenné dučeje, po němž se výrazně zmenší plicní průtok. Hypoxické záchvaty s těžkou cyanózou a hyperpnoí se vyskytují u neoperovaných dětí při protrahované desaturaci, vznikající následkem zvýšených kontrakcí infundibula pravé komory. Při těchto hypoxických záchvatech si děti dřepají, tím dojde ke zvýšení žilního návratu i systémové vaskulární rezistence a následně ke zmenšení pravo-levého zkratu. Farmakologicky lze záchvat potlačit podáním beta-blokátorů na principu snížení kontraktility myokardu. Chronická cyanóza vede k sekundární erytrocytóze (polyglobulii). Typickým příznakem jsou i paličkovité prsty s nehty tvaru hodinových sklíček. Vzácně existují i nemocní s mírnou formou Fallotovy tetralogie s lehkou pulmonální obstrukcí a malým nasedáním aorty, kteří mohou být klidově téměř bez cyanózy a mohou se dožít dospělého věku bez operace a bez větších obtíží (tzv. „růžový Fallot“).
DIAGNÓZA
Poslech: hlučný systolický šelest crescendo-decrescendovitého charakteru s maximem ve 3. mezižebří vlevo parasternálně (nad infundibulem pravé komory).
Pohled: centrální cyanóza, paličkové prsty s nehty tvaru hodinového sklíčka.
EKG: izolovaná hypertrofie pravé komory.
RTG srdce a plic: Zvednutý hrot při hypertrofii pravé komory a konkavita v místě plicnice (hypoplazie plicnice) dává srdci typický tvar („coeur en sabot“ nebo "dřevákovitý tvar"). Plicní cévní kresba je chudá z důvodu sníženého plicního průtoku.
Echokardiografie: základní a hlavní diagnostická metoda. Kromě stanovení diagnózy vyloučí i jiné přidružené vady a umožní diferenciální diagnostiku cyanotických srdečních vad.
Katetrizace: její význam poklesl s rozvojem echokardiografie. Katetrizace i v dnešní době upřesňuje morfologii výtokového traktu pravé komory, plicnice a jejích větví, morfologii a odstupy koronárních tepen, zobrazuje vlastní aortopulmonální kolaterály, před operací jsou důležitá hemodynamická data. Pomáhá lépe zobrazit morfologii velkých cév po předchozích spojkových operacích.
OPERACE
Kojenci s cyanózou a těžkými hypoxickými záchvaty nereagujícími na farmakoterapii jsou pro kritickou vadu indikováni k operaci. Pokud mají dobře vyvinuté plicní cévní řečiště a nemají hypoplastickou levou komoru, přistupujeme k radikální korekci vady v 6 - 12 měsíci. Při hypoplázii plicních cév nebo levé komory se nejprve provádí arteriální spojková operace. Jakmile se po navýšení plicní cirkulace zlepší kapacita plicního řečiště a levé komory, je možné v druhé době přistoupit k definitivní korekci vady.
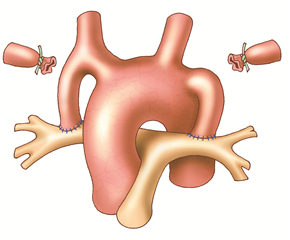
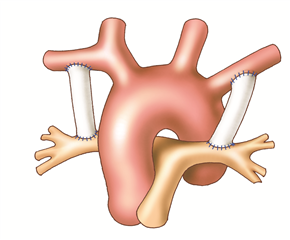 Principem paliativních spojkových operací je přivést do plic více smíšené krve ze systémového oběhu. Nejznámější je subklaviopulmonální spojka dle Blalocka-Taussigové, při které se sklopí konec podklíčkové tepny ke straně plicní tepny. Nyní se více užívá tzv. modifikovaná Blalockova spojka, kdy spojení mezi podklíčkovou tepnou a větví plicnice je zajištěno pomocí interponované goretexové protézy. Provádí se z pravo- nebo levostranné torakotomie v závislosti od výběru podklíčkové tepny (preferovaná je pravá). Další možností je vytvoření tzv. centrální spojky dle Amata (spojení vzestupné aorty a kmene plicnice protézou) z levostranné torakotomie nebo mediánní sternotomie.
Principem paliativních spojkových operací je přivést do plic více smíšené krve ze systémového oběhu. Nejznámější je subklaviopulmonální spojka dle Blalocka-Taussigové, při které se sklopí konec podklíčkové tepny ke straně plicní tepny. Nyní se více užívá tzv. modifikovaná Blalockova spojka, kdy spojení mezi podklíčkovou tepnou a větví plicnice je zajištěno pomocí interponované goretexové protézy. Provádí se z pravo- nebo levostranné torakotomie v závislosti od výběru podklíčkové tepny (preferovaná je pravá). Další možností je vytvoření tzv. centrální spojky dle Amata (spojení vzestupné aorty a kmene plicnice protézou) z levostranné torakotomie nebo mediánní sternotomie.
Radikální operace Fallotovy tetralogie spočívá v uzavření subaortálního defektu komorového septa záplatou, dále v uvolnění stenózy plicnice a rozšíření zúženého výtokového traktu pravé komory. Provádí se v mimotělním oběhu s kardioplegickou srdeční zástavou přístupem ze střední sternotomie. Intrakardiální výkon se provádí skrz pravou síň a truncus pulmonalis (transatriálně-transpulmonální přístup). Nahradil původní transventrikulární přístup, který zvyšoval riziko arytmií a dysfunkce pravé komory v dlouhodobém pooperačním průběhu. V případě, že je nutná intervence na hypoplastickém anulu pulmonální chlopně, používá se k rozšíření perikardiální záplata, někdy s uměle vytvořenou perikardiální chlopní. Stejně ale pooperačně vzniká různě závažná pulmonální regurgitace. Mírná není prognosticky závažná, ale významná regurgitace vede po letech k selhání pravé komory na podkladu objemového přetížení. V těchto vzácných případech je nutno stav řešit implantací nejlépe biologické chlopně do pulmonálního ústí.
VÝSLEDKY
Operační riziko se v současnosti pohybuje pod 1 %. Prognóza po radikální operaci Fallotovy tetralogie je dobrá. Sledování kardiologem je nutné z důvodu různych komplikací: rekanalizace defektu, reziduální obstrukce výtokového traktu pravé komory, pulmonální chlopně nebo plicnice, pulmonální regurgitace a pravostranné srdeční selhání, dysrytmie (z okolí jizvy po ventrikulotomii), aneuryzma pravé komory v okolí ventrikulotomie, infekční endokarditida.
Transpozice velkých cév
 MORFOLOGIE
MORFOLOGIE
Za tuto anomálii je považována především tzv. nekorigovaná transpozice velkých tepen. Srdeční dutiny jsou uspořádány v atrioventrikulární konkordanci a odstupy velkých tepen ve ventrikuloarteriální diskordanci (velké tepny tedy odstupují z nepříslušných komor - aorta z pravé komory a plicnice z levé komory). Asi polovina až 2/3 nemocných má izolovanou (prostou) transpozici, bez přidružených vad, zbylé vady jsou komplexní, nejčastěji spojené s defektem komorového septa nebo s obstrukcí výtokového traktu levé komory (subpulmonální stenózou). Nekorigovaná transpozice velkých tepen znamená dva nezávislé oběhy, které bez vzájemné komunikace nejsou slučitelné se životem. Po narození zajišťuje komunikaci po několik dnů otevřené foramen ovale a otevřená tepenná dučej (a taky případný defekt komorového septa). Jakmile se přirozené zkraty uzavírají, vada rychle vede ke kritické hypoxémii a úmrtí dítěte.
Při zachovaných zkratech dochází k hypertrofii pravé komory, která se adaptuje na systémový tlak. Levokomorový myokardu naproti tomu zůstává slabý vzhledem k nízkému odporu plicního řečiště.
SYMPTOMY
Příznaky se objevují hned po narození a závisí na konkrétní morfologii vady. Vada se chová vždy kriticky. Objevuje se především cyanóza, tachypnoe, dyspnoe, tachykardie a městnavé srdeční selhání.
DIAGNÓZA
Poslechový nález bývá chudý.
EKG: známky hypertrofie pravé komory.
RTG srdce a plic zobrazí patrné zvětšěné srdce vejčitého tvaru s úzkou stopkou a výraznou plicní cévni kresbou.
Echokardiografie: suverénní diagnostická metoda. Zobrazí morfologii vady, odstupy velkých tepen a koronárních artérií z aorty, velikost i tvar komor, ev. i další přidružené anomálie.
Katetrizace není pro stanovení dianózy nutná, umožňuje ale život zachraňující intervenci - balonkovou atrioseptostomii.
OPERACE
U prosté transpozice je po narození indikována urgentní balonková atrioseptostomie dle Rashkinda a podání prostaglandinu E1 – pro zachování přirozených zkratů v cirkulaci (foramen ovale a ductus arteriosus). Do 2, nejpozději 3 týdnů života je nutno provést anatomickou korekci vady dle Jatena (ještě v době, než levá komora odvykne tlakové zátěži). U kojenců, u nichž v novorozeneckém věku nebyla provedena časná anatomická korekce, je nutno levou komoru před anatomickou korekcí "natrénovat" provedením bandáže plicnice a současně provedenou spojkovou operací (cévní protézou se spojí levé podkličková arterie s levou větví plicnice). Po 7-10 dnech tuto již tlakově zatíženou levou komoru lze zapojit Jatenovou operací do systémové cirkulace. U pozdně diagnostikované transpozice je natrénování možno provádět i později, vyžaduje to ale delší časový interval (až do 1 roku).
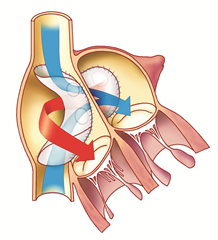
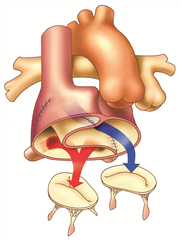
Anatomická korekce vady (Jatenova operace, arteriální switch) je radikální výkon na úrovni velkých tepen, při kterém se protnou a vymění aorta s plicnicí a koronární tepny se reimplantují do neoaorty. Preferovaný postup.
Fyziologické korekce vady (dle Mustarda nebo dle Senninga) jsou výkony na úrovni síní, v současnosti se u nekorigované transpozice již neprovádějí. Principem je nasměřovat všechnu žilní krev přitékající do pravé síně horní i dolní dutou žilou do levé komory, ze které odstupuje plicnice. Krev z plicních žil je kompletně derivována cestou levé síně do pravé komory a do aorty. Toho lze dosáhnout zkonstruováním tunelu uvnitř síní z perikardu (Mustardova operace) nebo využitím stěny pravé síně (Senningova operace).
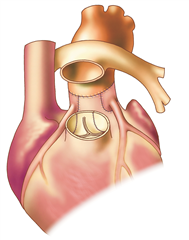
VÝSLEDKY
Operační mortalita prosté transpozice při fyziologické korekci se dnes pohybuje kolem 3 % a při anatomické korekci dokonce pouze mezi 1 - 2 %. Bez operace zemře 90 % dětí v průběhu prvního roku života. Operovaní mají celkem dobrou další životní prognózu. V dospělosti se však může po fyziologických korekcích rozvinout dysfunkce pravé (systémové) komory s významnou trikuspidální insuficiencí, dále dysrytmie, stenózy vústění horní nebo dolní duté žíly, obstrukce plicních žil. Po anatomické korekci je obávanou komplikací především ischemie myokardu ze zalomení nebo ze stenóz ústí implantovaných věnčitých tepen a také rozvoj insuficience chlopně neoaorty.
Zdroje:
- Hučín B. Dětská kardiochirurgie - 2., doplněné vydání. Praha: Grada, 2012. ISBN 978-80-247-4497-1.
- Češka R a kol. Interna. Praha: Triton, 2010. ISBN: 978-80-7387-423-0.
- Dominik J. Kardiochirurgie. Praha: Grada Publishing, 1998. ISBN: 80-7169-669-2.
- Dominik J.: Kardiochirurgie. In.: Pafko P. et al.: Základy speciální chirurgie, Galén, 2008, s.173-195. ISBN 978-80-7262-402-7
Za konzultaci a aktualizaci dat děkujeme MUDr. Romanovi Gebauerovi, primáři Kardiochirurgického oddělení Dětského kardiocentra Fakultní nemocnice v Motole v Praze.
